REGULATORY
Access to Marketing Authorisation
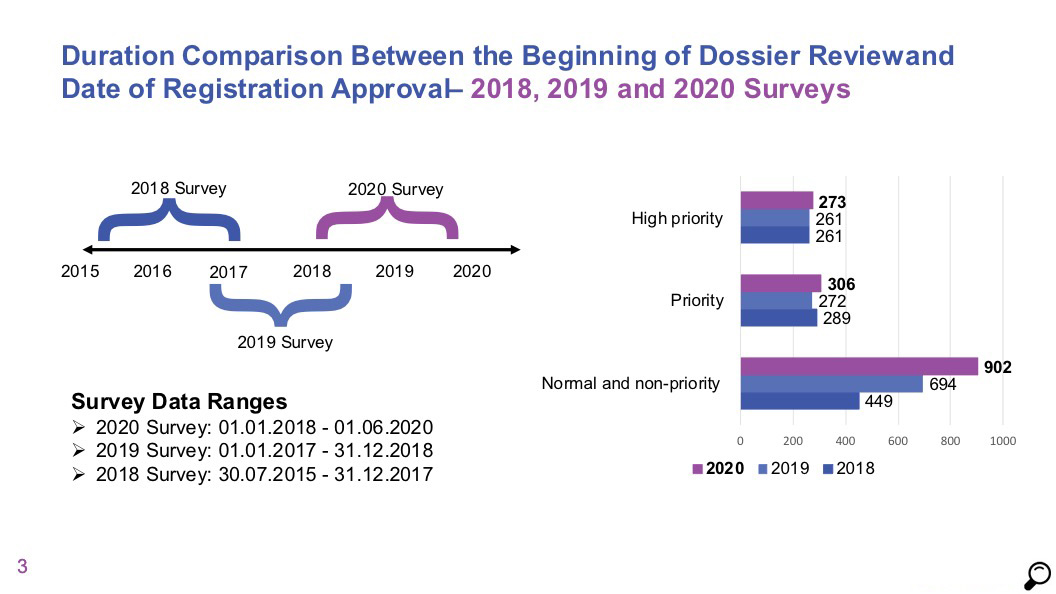
According to the data of AIFD 2020 survey covering the period between 01.01.2018 and 01.06.2020;
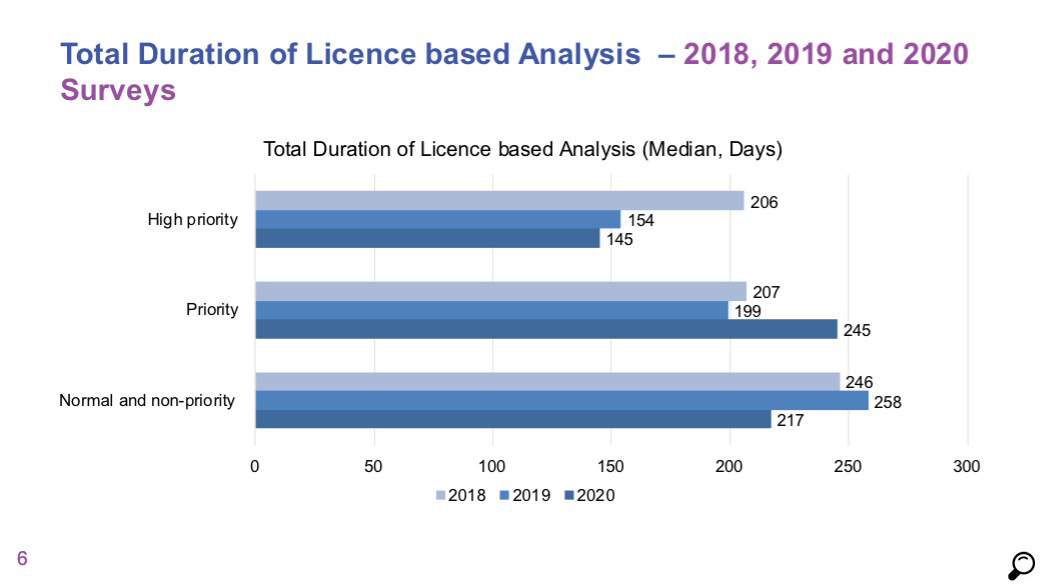
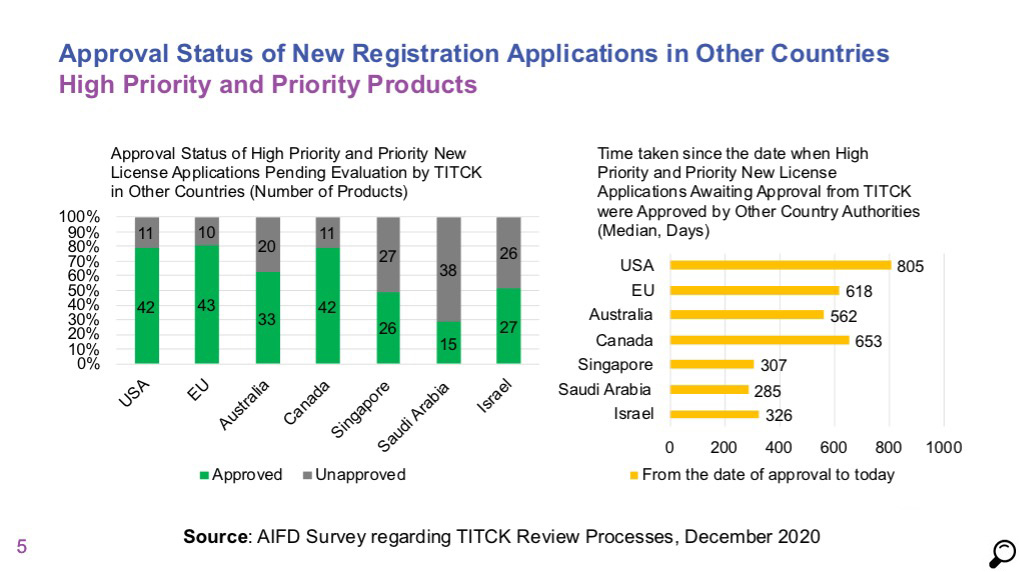
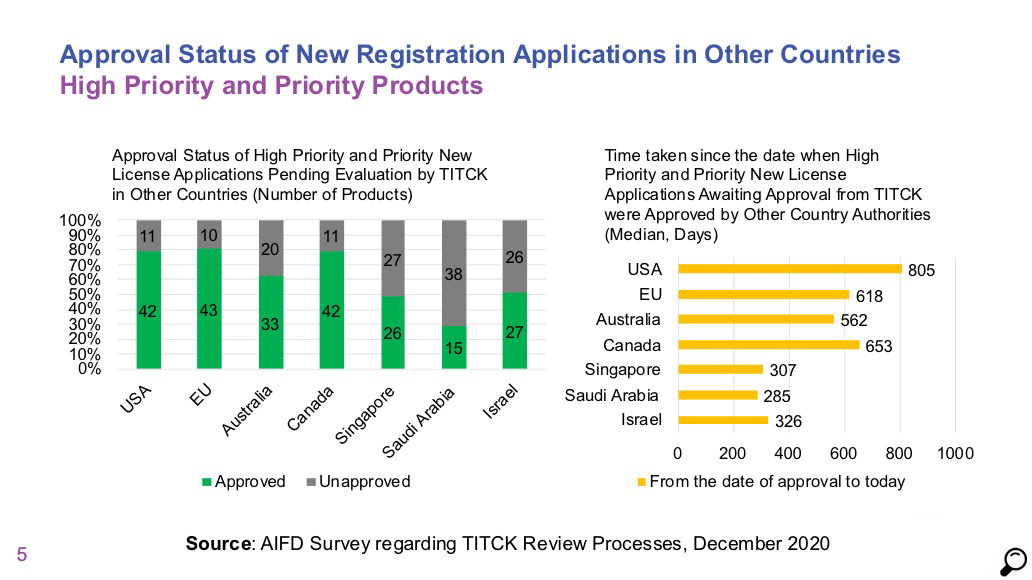
- Comparison of licensing timelines of high priority and priority products in 2018 and 2019 data does not show a significant change. Median duration for high priority products was 273 days and it was 306 days for priority products.
- For normal priority / non-priority products, the median duration was measured as 902 days, which indicates that no progress has been made for products in this category compared to 2018 and 2019 data, and that the process is getting longer.
 Prioritisation Process
Prioritisation Process
One of the studies carried out within the scope of "Accelerating Access to Innovative Medicines" was AIFD's opinions and suggestions submitted to TITCK in 2018 for the revision of the guideline on the working procedures and principles of the Human Medicinal Products Priority Assessment Board.
Though there was no development in 2019 regarding the guideline revision, a 2-year estimated financial impact projection is now started to be requested during the prioritization applications for innovative drugs. Several problems may stem from such an approach:
- According to the registration regulation, in order to be licensed, the effectiveness, safety and quality of a product must be proven and a file prepared without any cost information is presented during the application. A prioritization assessment during the licensing process made according to the effect of the product on public finances takes the process out of the medical / scientific evaluation framework. In the regulatory process, which includes a clinical and technical evaluation based on scientific data only, the cost of an innovative drug should not be considered as an influencing factor.
- At the time of prioritization application, the price of the product may not yet be determined / published in many countries. In addition, when new treatments are first introduced to the market, they may be marketed in higher priced countries other than reference countries, causing the source price to appear higher than it would be. For this reason, even if the price is available, companies may postpone their applications for innovative treatment, as the reliability and validity of this price will be uncertain in the medium term.
- One of the price determining factors of a medicine is the market dynamics. At the GMP / license application stage, companies may not yet have the relevant information and / or the market dynamics may change while the process continues. For example, new studies / data on the disease may be published, other medicines may be withdrawn from or enter to the market, or there may be changes in the existing treatments as a result of new experiences or changes in treatment protocols.
- The market price of a medicine is established after the product is marketed in certain countries and after the first health technology evaluations; the treatment protocols are included in the guidelines at these stages. Therefore, it is very difficult at this stage for a company to have a comparable treatment protocol and cost data of the treatment.
AIFD has always advocated that the prioritization process should include clinical and technical assessment based only on scientific data,
independent of the price commitment and projected two-year estimated financial impact.
In the prioritization criteria of health authorities such as the American Food and Drug Administration (FDA), the European Medicines Agency (EMA) and the Canadian Agency for Drugs and Technologies in Health (CADTH), the licensing processes are defined only by unmet treatment needs. When the practices and regulatory pathways used by authorities of the developing countries to prioritize "the unmet need for treatment" are reviewed, the economic criteria of prioritization assessments such as impact on public finance and on the current account deficit, do not appear to be used by any other country than Turkey.
Based on the premise that the main goal of the prioritization process is “to provide patients the fastest access to the best treatment”, the financial data requested in the applications will lead to a prolongation of patients' access to innovative treatments and to an increase in unmet treatment needs.
This issue was brought by the AIFD Board of Directors to the agenda at the meetings with the Ministry of Health, with the Deputy Minister of Treasury and Finance, with TITCK President and Vice President, and it was communicated in writing to all stakeholders. It was emphasized that these data requested in the applications may lead to the prolongation of the patients' access to innovative treatments and an increase in the unmet treatment need.
Moreover, in a survey conducted AIFD member companies it was observed that;
- There are innovative products for which GMP prioritization application has not been made since the price has not been determined yet, and parallel license applications cannot be made because these products cannot be prioritized.
- Many of these products are currently considered subject to accelerated / priority licensing processes by the European Union and the US health authority.
- Currently 24 clinical trials are ongoing in Turkey with these products for which prioritisation application cannot be made. It is believed that the prolongation or uncertain registration periods that will occur during the licensing processes of these products would also have a negative impact on their new clinical trial investments.
Reducing the time gap between the first registration approval times in the world and in our country is extremely important, especially in terms of accelerating the access of patients to innovative medicines. However, the price commitment letter and newly requested financial projections included in the Guidelines for Human Medicinal Products Priority Assessment Board Working Procedures and Principles affect both the prioritization and license application time, and will lengthen not only the license approval periods but also the time gaps between the application periods. As a result of the prolongation of GMP and licensing processes, there is a risk that, as in the previous years the number of medicines procured from abroad through TEB will increase to allow patients to access innovative treatments.
It is assessed that taking prioritization decisions based on the impact of innovative products on public finance would also disrupt the Pharmaceuticals and Medical Devices Agency's vision to become a reference health authority accepted on a global scale and would delay patients access to innovative treatments. Our advocacy is ongoing within this framework to stop this approach as soon as possible.
Analysis for Licensing
Another action under the “Acceleration of Access to Innovative Pharmaceuticals” goal was towards the improvement of analysis processes at the licensing stage.
Although the analysis process is run in parallel with the other assessment and licensing evaluation processes, the analysis process itself is taking a long time and various issues experienced in the processing delay license approvals. This delay has lately been experienced more acutely in high technology biotechnology products and in blood products.
Basically, as issues experienced in the analysis- for-licensing process causes delays in licensing, there is a need to make the relevant law and regulation amendments in order to conduct the analysis within the framework of market controls as a separate process, without hindering the licensing activity.
A “TITCK & AIFD Workshop on Analysis For Licensing” was held on December 18, 2019 in Ankara to share the issues and our proposed solutions with TITCK officials on a resolution of topics during the transition period until the suggested changes to laws and legislations are completed.
Our expectations and proposed solutions communicated to the Institution’s officials during the workshop:
- In the draft regulation, inclusion of the process of analysis for licensing to the 210 days licensing timeframe;
- As companies are notified about deficiencies of analysis only at the late stage of licensing, product registrations are delayed; there is a need for harmonization of intra-institutional processes on the communication on analysis needs;
- Development of interim solutions for the supply of samples and materials and for vitally important products such as biological/biotechnological products until the suggested changes to laws and legislations are made
The Indication Approval Processes of Innovative Medicines
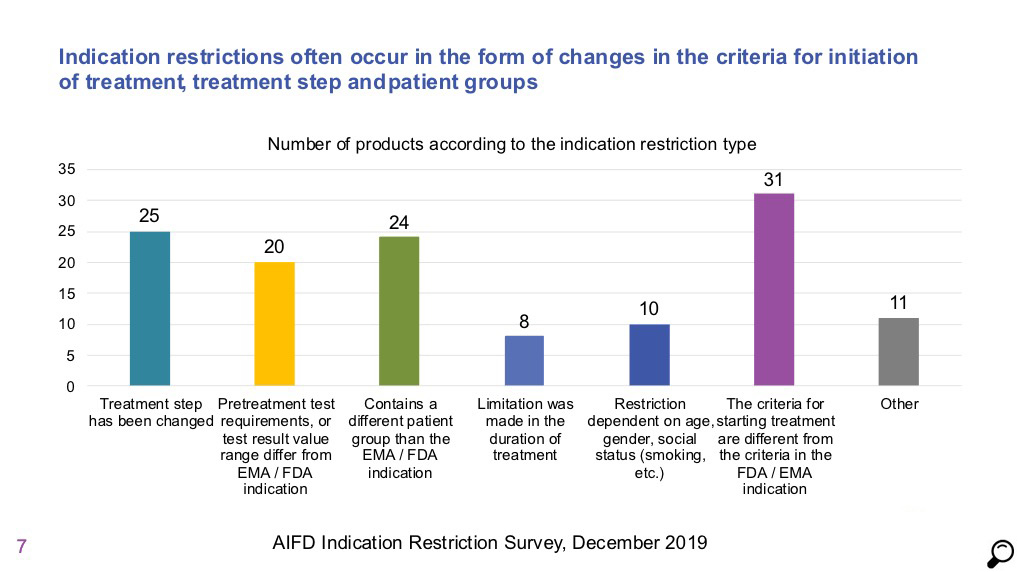
Another barrier against “Access to Innovative Medicines” is the frequent indication limitations imposed specifically in the assessment of FDA and EMA conform applications for indications specially in oncology, hematology, respiratory diseases and immunology products.
As such, assessment processes for indication has been an area that has challenged us the most in recent years. Cases such as issuing a common indication in the indication assessment process for products that are indicated under the same field of treatment irrespective of the clinical trials conducted with the product and, adding reimbursement conditions/criteria on indication texts during the process of scientific assessment result in products to be approved with indications incompatible with clinical studies on which they are based.
This situation has negative outcomes such as causing delays in the market introduction of innovative products or decisions to not launch these products in the Turkish market as the scientific literature created for these products cannot be used in their promotion to healthcare professionals.
It is critical that in the indication approval process the indication assessments should be purely based on scientific data without factoring in the budgetary impact or pharmaco-economic evaluations, in order to permit patient access to the right and effective treatment.